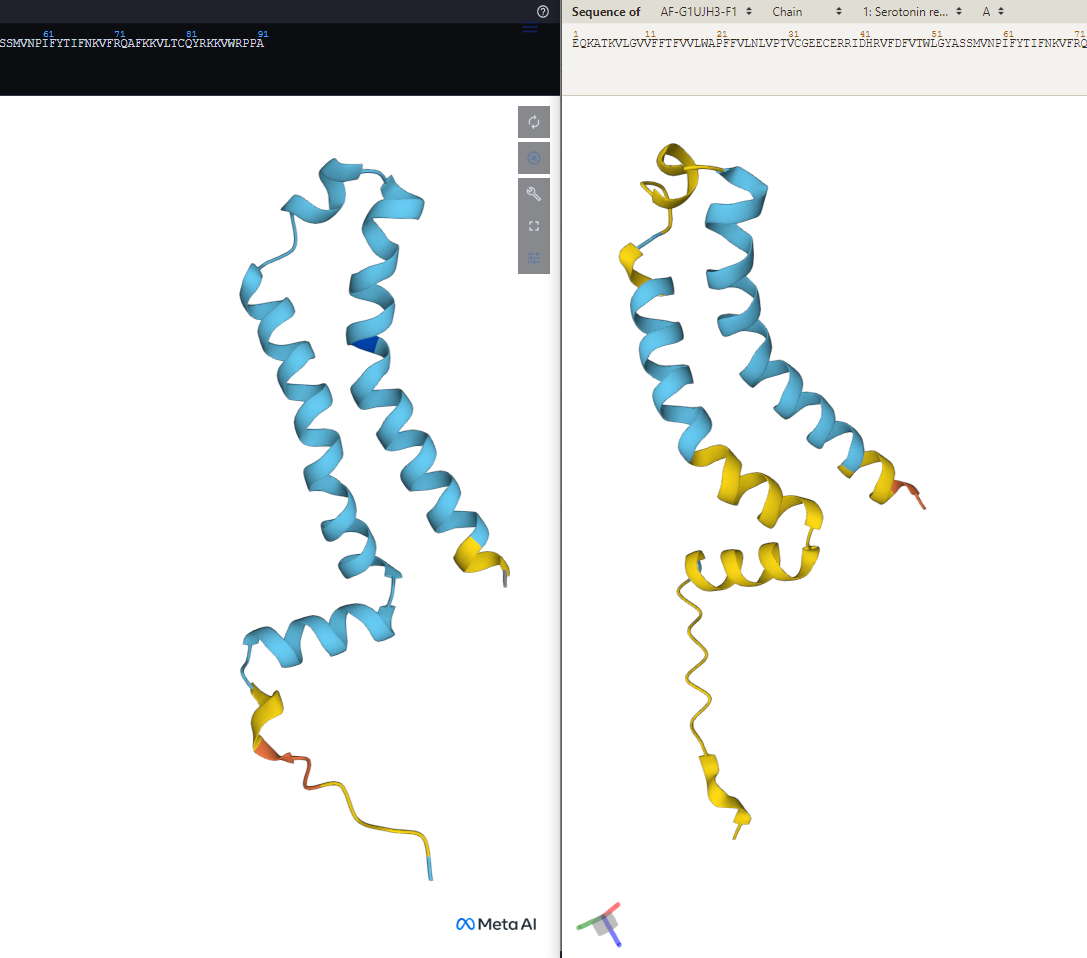
 Shape of a protein predicted by two different AI models (ESMFold on the left, AlphaFold on the right)
Shape of a protein predicted by two different AI models (ESMFold on the left, AlphaFold on the right)wazabee t1_iw2up7u wrote
Reply to comment by fuckpudding in Shape of a protein predicted by two different AI models (ESMFold on the left, AlphaFold on the right) by greentea387
Very. Proteins must maintain their shape as much as possible if they are function. When a protein interacts with another compound or structure, the positioning of the amino acid side chains is crucial. The side chain positioning acts lkle a key in a lock, and exert their influence based on their angle, distance, and positioning relative to what they interact with.
NineMinded t1_iw3avte wrote
While this might be true, getting close to the correct shape can speed research, I assume.
wazabee t1_iw3jc6d wrote
For research purposes, this is perfect. It's the best approximation to the native protein structure we can get. Before this, we had xray crystallography, but the problem with that was it cause the protein to change shape.
Money_Cut4624 t1_iw5k3tj wrote
It's just in silico model, it needs a whole process to test a protein in vitro.
wazabee t1_iw5qi2v wrote
Just combine this with a 3D NMR and cryo EM model and you're good to go.
styxboa t1_iw9u0e8 wrote
What's that? can you explain this concept to me and why it'd help
wazabee t1_iw9w2bk wrote
the 3 dimentional structure of a protein is key to its function, so to understand what a particular protein does or how disease occurs, we want to look at it. We have multiple different methods to do this, and each has their advantages. Our main goal when looking at a protein is to see it in its original form, as would be found in the cell, otherwise known as the native structure. The issue with some techniques, such as x ray crystallography, is that the conditions required to cause a protein to crystalize lie outside the normal function range the protein works in, so the shape we see is more of an approximation. 3D NMR is a technique that is capable of seeing a molecule based on how atom react to an external magnetic field. With 3D NMR we are looking at this reactivity using 3 atoms: hydrogen, carbon, and nitrogen. From the gathered data, we can form a 3d computer model that is more closly resembling the native structure. The additional advantage of 3D NMR is that we can see the areas of the protein that are static and other parts that are more dynamic. Cryo EM or cyrogenic electron microscopy requires us to freeze the proteins on a platoform, and then send a beam of electrons to see the proteins. The resulting image is a blurred representation of the protein, but acts as a quick and easy starting point for research. When combined together, we get a good idea of what the structure looks like.
Xray crystallography produces the most detailed images we can get, up to the resolution of 1 angstrom, which lets us see hydrogen bonding, but getting the protein and having it synthesized in a manner that allows it to be imaged is just one of the few headaches researchers have to go through before they can get an image. When I was a researcher the lab next to us worked with cryo EM. It was cool.
cat3cat123 t1_iw6bw1r wrote
To be fair, this modeling is based a lot off of learning from previous crystal structures and the biases those may or may not impose. An x-ray crystal model (or NMR/Cryo-EM structure if you have a very small or very large protein respectively) is still considered the “ground truth”, while these machine learning generated structures are more for hypothesis generation/approximations (that may help in building models from experimental structural data).
Crystal packing needed for x-ray crystallography may rigidify a protein, but at least within the field of structural biology it is not believed to alter the shape of the protein. Likely it just hides the dynamic conformations a protein can occupy - and machine learning methods also suffer from this flaw as they only predict a single structure.
vhu9644 t1_iw6folh wrote
Really? Could these both not be viable structures that a protein could switch between due to thermal fluctuations?
It looks like it’s not a particularly complex protein, so I imagine it’s some ligand or subunit for something, in which case the “correct” structure would be stabilized by its interaction with another object.
wazabee t1_iw7a1gw wrote
Protein structures have both static and dynamic sections inside of them, but computer models are not not well suited to predict them. You'd need to confirm you findings with an imaging experiment, like 3d NMR, that is capable of capturing those dynamic, shifting structures.
vhu9644 t1_iw7c7do wrote
Sure, but I’m just skeptical of the claim that these two predicted structures would give wildly different functions, or that they really are distinct on something this simple.
I cold believe it if for example the catalytic core of a barrel protein had small alterations in structure, but this is just two helices next to each other with a small disordered domain on the bottom.
wazabee t1_iw7cgjz wrote
It can. A single point mutation of one amino acid is enough to cause a deviation in normal function, as is the case of some diseases.
vhu9644 t1_iw7ct55 wrote
Yes I’m aware.
But these arguments by analogy don’t do it for me for something this simple, that doesn’t even look like it would have a catalytic core without some other subunit. Do you even know what protein this is?
Edit:
It’s a serotonin receptors from cricket. It’s a membrane protein so it should be stabilized by going through the membrane.
wazabee t1_iw9uoon wrote
im a biochemist, so yes i know what a protein is. Im trying to explain the importance of maintaining the 3d structure of a protein to its function. Both proteins have the same sequence, but there is a slight difference in what the software rendered. Now, based on the sequence, a cell would reliably recreate the same protein. However, in the context of having 2 slightly different structures from the same amino acid strand, yes, you can have widely varying functions that result depending on the original purpose of the peptide strand.
vhu9644 t1_iwau2w1 wrote
And I’m a synthetic biologist in protein engineering. What I’m skeptical about is that for this protein specifically, this change in structure plays a major role in function determination, due to its simplicity, and that we are seeing two distinct folds that are locked from each other.
The point ultimately is moot, the protein chosen is a membrane bound protein, so the lipid layer will provide stabilization.
Viewing a single comment thread. View all comments